固体中的原子结合¶
- 原子为什么能够结合成稳定的固体?
- 不同结合方式有何物理差异?
密堆积和配位数¶
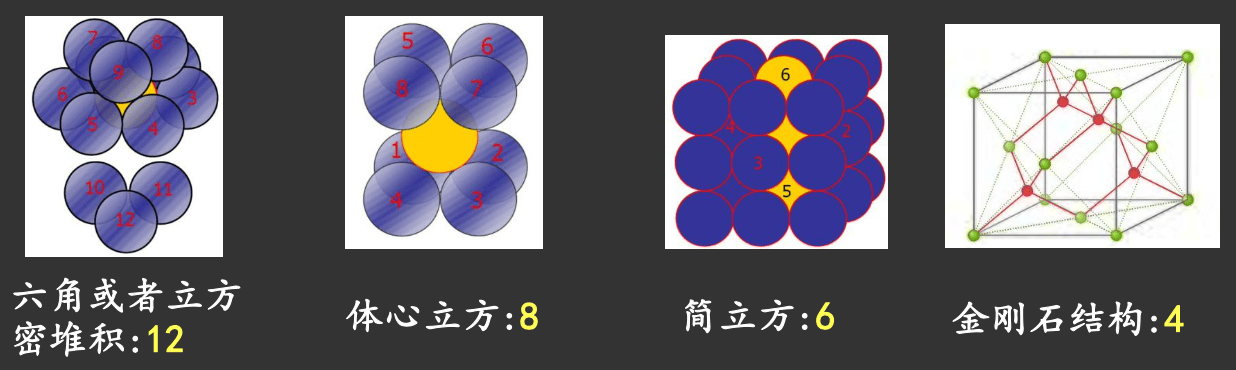
在一定温度下,原子在晶体中总是尽可能地通过紧密排列来降低体系自由能,而简单情况下原子可看作等体积刚性球。
 如何最紧密堆积?\(\implies\) Kepler 堆积问题(1611)
如何最紧密堆积?\(\implies\) Kepler 堆积问题(1611)
配位数:一个原子的最近邻原子数
最大配位数(密堆积):\(12 = 6 + 3 + 3\)
由对称性限制,可能的配位数是 \(12, 8, 6, 4, 3, 2\)
能量角度理解晶体的形成¶
结合能 \(W\):自由粒子体系与晶体体系的能量差
离子性结合¶
- 基本特征:通过电子转移形成带电离子,以离子作为结合单元构成晶体
Evjen 晶胞法:近邻、次近邻、次次近邻...
\(\mathrm{NaCl}\) 的马德隆常数 \(\alpha = 1.748\)
原子间距 \(\downarrow\),晶体体积 \(\downarrow\),电子整体密度 \(\uparrow\),电子云交叠,产生排斥作用,体系能量升高。
排斥势难以由第一性原理直接导出,故采用经验形式
体系内能
- 重叠排斥为短程相互作用,随距离变化极为陡峭(\(n \gg 1\))
平衡条件和稳定条件¶
-
平衡条件:合力为零
\[ f(r_0) = -\left. \frac{d u(r)}{d r} \right|_{r_0} = 0 \implies B = \frac{\alpha e^2}{4 \pi \varepsilon_0 n} \frac{r_0^{n - 1}}{n} \]结合能
\[ W = -Nu(r_0) = \frac{N \alpha e^2}{4 \pi \varepsilon_0 r_0} \left( 1 - \frac{1}{n} \right) \]由于 \(n \gg 1\),所以结合能主要由库伦吸引决定(排斥项只在短程起作用)
-
稳定条件
共价结合¶
- 基本机制:两个原子各提供一个电子(自旋相反),形成成对电子态(bonding state)
- 核心特征:“饱和性”
- 每个原子可形成的共价键数目存在上限
-
若价电子数 \(N \geq 4\),则
\[\text{键数} = 8 - N\]成键满足“八隅体规则”:8 个价电子 - 若价电子数 \(N < 4\),则
\[\text{键数} = N\]成键后相对稳定,是 局部 最低能量状态 - 方向性:共价键只沿特定方向形成 - 来源于轨道空间结构
能量角度理解共价键的形成¶
- 原子接近 \(\to\) 波函数重叠 \(\to\) 形成新的量子态
- 电子成对(自旋相反)占据低能态 \(\to\) 能量降低 \(\to\) 成键
降低的能量即为成键能
\(\mathrm{H}_2\) 分子:两个原子核 \((a, b)\) + 两个电子 \((1, 2)\). 此为四体问题(Four-body problem)!无法解析求解,采用近似方法:分子轨道法
分子轨道法
原始多体哈密顿量,绝热近似,认为原子核不动,忽略了原子核的动能和相互势能(为常数)
因为 \(V_{12}\) 的耦合,上面的哈密顿量无法对角化严格求解,故将其忽略,得到单电子薛定谔方程
电子在两个原子核形成的有效势场中运动,\(\psi_i(\mathbf{r}_i)\) 称为分子轨道波函数.
分子轨道的构造:LCAO 方法
分子轨道描述电子在整个分子范围内的概率分布,而不是局域在某一个原子上。
-
基本假设 分子轨道可以表示为原子轨道的线性组合(Linear Combination of Atomic Orbitals, LCAO):
\[ \psi_i(\mathbf{r}) = \]
概率密度与成键/反键的本质
概率密度表达式
- 成键态:干涉项为正,中间电子密度增加
- 反键态:干涉项为负,中间电子密度减少
成键本质:电子分布在原子核间区域概率密度的增强
概率密度的演算
设核点中点为 \(x = 0\),两个 1s 轨道到中点的距离为 \(R/2\),则
能量的升降来源于电子在原子核间区域的重新分布。电子在核间时,同时靠近两个原子核,所以势能项 \(V = -\frac{e^2}{r_a} - \frac{e^2}{r_b}\) 更小,势能显著降低(主要);电子动能相比于两个孤立原子中的电子会略微上升(次要)。
成键态电子动能增加是由于 Pauli 不相容原理导致的吗?
否。 是波函数的空间结构变化引起的!
是波函数的空间结构变化引起的!
电子气/约束多电子体系,电子密度增加导致被填充的态变多,动能增加,这是 Pauli 不相容原理引起的。
但是单个波函数,不存在被迫占据高能态的问题!那么波函数的局域程度变化使动能升高的原理是什么?
Heisenberg 不确定性原理!\(\Delta x \downarrow \implies \Delta p \uparrow \implies T \uparrow\)
对于反键态,动能显著增加!从波函数的空间局域理解:弥散的波函数随空间的变化小,而局域的波函数随空间的变化大。
局域强,梯度大,动能显著上升。同时势能也增加了:
核间电子被挤到两边,中间电子密度几乎为零
| 性质 | 成键态 | 反键态 |
|---|---|---|
| 波函数 | 同相相加 | 反相相加 |
| 核间区域 | 电子密度增加 | 电子密度减少 |
| 动能 \(T\) | 略微增加 | 显著增加 |
| 势能 \(V\) | 显著降低 | 升高 |
| 总能量 | 降低(稳定) | 升高(不稳定) |
从原子轨道到共价成键:以金刚石为例
碳原子基态电子组态:\(1s^2 2s^2 2p^2\),只有 2 个未成对电子,但为什么金刚石可以和周围 4 个碳原子形成共价键?
 杂化轨道电子组态:\(1s^2 2s^1 2p^3\),形成四个等价轨道!
杂化轨道电子组态:\(1s^2 2s^1 2p^3\),形成四个等价轨道!
有一个 \(2s \to 2p\),能量略微升高!成键降低的能量必须弥补这个升高的能量。
-
处于杂化轨道状态的电子平均能量成为杂化能:
\[ \varepsilon_h = \frac{1}{4} (\varepsilon_{s} + 3 \varepsilon_p) \] -
增加的能量称为晋升能(promotion energy)
-
成键后,体系能量的降低称为成键能(bond energy)
- 当原子距离过近时,短程排斥相互作用会迅速增强,使体系能量升高 \(\phi(d)\)
综上,总的结合能
化学键研究的先驱
Linus Carl Pauling (1901-1994),1954 Nobel 化学奖,1963 Nobel 和平奖
杂化轨道理论认为,在形成化学键的过程中,原子轨道本身会重新组合,